Этиология
Этиология псевдогипертрофической формы Дюшенна.
Псевдогипертрофическая прогрессирующая мышечная дистрофия (мышечная дистрофия Дюшенна, Xp21.2, ген DMD дистрофина) — возникает в результате дефектов гена, кодирующего белок дистрофин. Дистрофин локализован в плазматической мембране скелетных мышечных волокон и кардиомиоцитов. Мутации в гене дистрофина вызывают клинически явно различающиеся формы миодистрофий [Евтушенко С.К., Садеков И.А. 1994].
Некоторые авторы предполагают, что различия в клинике дистрофинопатий могут определяться характером мутаций, из которых одни приводят к сдвигу рамки считывания (в результате чего синтез соответствующего белка практически невозможен), тогда как другие повреждают ген, но не нарушают рамку считывания (результатом чего становится синтез измененного белка, частично способного к функционированию) [Евтушенко С.К., Садеков И.А. 1994]. Первый вид мутаций обычно связывают с тяжелым течением болезни, второй - с более мягким. Данное предположение, известное как гипотеза Монако, нашло подтверждение в ряде публикаций [Гринио Л.П., Агафонов Б.В., 1997].
Вместе с тем накопление сведений о мутациях в гене дистрофина и изучение клинико-генетических корреляций при дистрофинопатиях способствовали выявлению случаев, трудно объяснимых с позиций гипотезы Монако или других гипотез, постулирующих жесткую связь особенностей клиники с глубиной повреждения функции белка. Еще в 1988 г. было опубликовано сообщение о нескольких больных, у которых выявлялись делеции экзонов 3-7 гена дистрофина и соответственно сдвиг рамки считывания, но при этом заболевание протекало в мягкой форме - как миодистрофии Беккера [Шишкин С.С., 1997]. Даже если бы у пациентов только отсутствовал данный участок последовательности дистрофина (не говоря уже о нарушении рамки считывания для остальной части гена), можно было бы ожидать серьезного нарушения функции белка, так как, по имеющимся сведениям, именно эта часть молекулы дистрофина обеспечивает его связывание с актином [McKusik V., Amberger J., 2003]. Однако болезнь в ряде случаев протекала относительно доброкачественно. Более того, известен пациент с делецией экзонов 3-9 в дистрофиновом гене, который до 60-летнего возраста и не подозревал о своей болезни, а в 67 лет сохранял способность к самостоятельной ходьбе [Ahn A.H., Kunkel L.M., 1998]. Авторы, описавшие этот уникальный случай, предположили, что, следовательно, возможны делеции в функционально важной области дистрофинового гена, которые обеспечивают состояние с длительным бессимптомным течением и без существенного сокращения продолжительности жизни [Kaplan J.C., Fontaine В., 1999].
В других публикациях отмечалось, что крупные делеции, захватывающие 26% (экзоны 21-44) и даже 40% последовательности дистрофинового гена, иногда обусловливают позднее начало и очень мягкое течение болезни - такие пациенты в возрасте 55 и 60 лет сохраняли определенную двигательную активность [Шаховская Н.И., 2000].
Этиология офтальмоплегической мышечной дистрофии Кило-Невина.
В 1998 г. изолировали на хромосоме 14q11.2-13 ген поли-(А)-связывающего белка 2 (PAPB2, PABPN1), ответственный за синтез ядерного белка PABP2, служащего фактором полиаденилирования мРНК, и идентифицировали мутацию, заключающуюся в увеличении числа копий тринуклеотидных GCG-повторов в 1-м экзоне гена. В норме ген содержит шесть тандемных копий повторов GCG, а у больных их число достигает до 8—13. В некоторых популяциях экспансия числа тринуклеотидных повторов происходит за счет простого добавления GCG-повторов, в других — вместе с экспансией GCG-повторов происходит GCA-вставка.
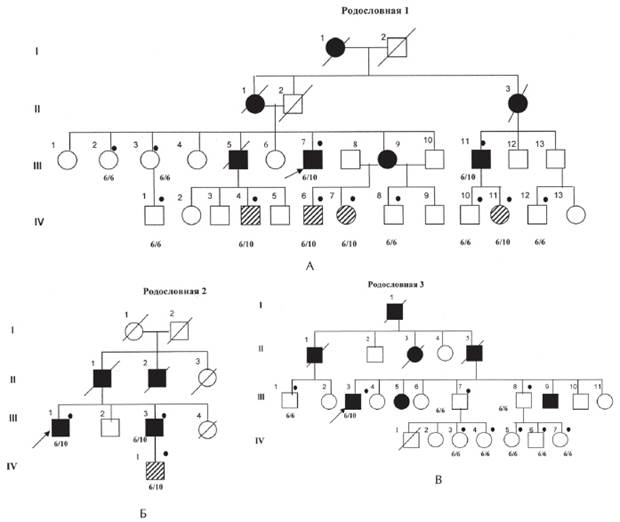
Рисунок 1. Рис. 1. А, Б, В. Родословные семей с офтальмоплегической мышечной дистрофии Кило-Невина с результатами ДНК-диагностики [Codere F., Brais B., Rouleau G., Lafontaine E., 2001].
Механизм развития экспансии тандемных повторов при офтальмоплегической мышечной дистрофии до сих пор неясен; высказываются предположения, что он может быть связан с неравным кроссинговером, разновидностью гомологичной рекомбинации, происходящей в зародышевых клетках во время мейоза или иногда митоза.
Также описаны случаи точечной мутации в гене PABPN1 22 и случаи гомозиготного носительства «промежуточного» аллеля гена, имеющего 7 GCG-повторов с аутосомно-рецессивным типом наследования с развитием более тяжелой клинической картины и более ранним дебютом симптомов заболевания [Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008].
Примечание. 6/6; 6/10 — результаты ДНК-анализа на мутацию в гене PABPN1; Закрашенный кружок —обследованные пациенты; закрашенный квадрат — больной с ОФМД, пустой квадрат — клинически здоровый; заштрихованный квадрат — клинически здоровый носитель мутации в гене PABPN1. I, II поколение — умершие родители, III поколение — больные и их сибсы, IV — дети, не достигшие возраста начала заболевания